|
|
|
|
Elements in nature come in forms called isotopes that differ only in the number of their neutrons. Most isotopes are stable and can be distinguished from their counterparts simply by their masses. Stable isotopes are denoted by their atomic mass such as 13C and 12C for the two stable isotopes of carbon, and 18O, 17O, and 16O for the stable isotopes of oxygen. Isotopes are species of a chemical element which differ from one another by atomic weight. The abundances of carbon and oxygen isotopes in nature are:
| Element | Heavy isotope | Light isotope |
| Oxygen | 18O = 0.2 % | 16O = 99.76 % |
| Carbon | 13C = 1.11 % | 12C = 98.89 % |

As shown by these equations, the δ value is a relative difference between a sample (RSample) and a standard (RStandard), and is a dimensionless parameter. Since values of δ in nature are small, they are generally given in ‰ (per mil) – which is a 103 factor, not a unit. Further, only the arbitrary choice of standard determines whether a δ value of a sample is positive or negative (Mook, 2000). The 18O/16O ratio (or 13C/12C ratio) is smaller in the sample than in the standard, then the sample is depleted in the 18O (or 13C, respectively) relative to the standard and the δ value is a negative number. Conversely, if the 18O/16O ratio (or 13C/12C ratio) is larger in the sample than in the standard, then the sample is enriched in 18O (or 13C, respectively) relative to the standard, and its δ value is a positive number. To allow efficient comparisons of results among different labs around the world, a series of common international standards has been established and all samples are normalized to such standards. For carbon, the common standard was originally a carbonate of the Pee Dee belemnite carbonate formation in South Carolina. Naturally, as the original standards are exhausted, appropriate substitutes have been introduced. The two main sources for international standards today are the International Atomic Energy Association (IAEA - https://www.iaea.org/) in Vienna, and the US National Institute of Standards and Technology (NIST - https://www.nist.gov/).
Within the field of physics, electromagnetic radiation embodies waves within the electromagnetic field, traversing space while carrying both momentum and electromagnetic radiant energy. The electromagnetic spectrum comprehensively encloses various forms of electromagnetic radiation, such as radio waves, microwaves, infrared, visible light, ultraviolet, X-rays, and gamma rays. Each segment of this spectrum engages in distinct interactions with matter, offering diverse applications in various scientific and technological fields.

Consider a white beam source, emitting light across multiple wavelengths, focused onto a sample, such as amorphic glass. When this beam interacts with the sample, photons matching the energy gap of the molecules present are selectively absorbed to excite these molecules. Simultaneously, other photons remain unaffected and transmit through. In cases where the radiation falls within the visible region (400–700 nm), the color of the sample becomes the complementary color of the absorbed light. Absorption spectroscopy serves as a vital analytical chemistry tool, adept at discerning the presence of specific substances in a given sample. Furthermore, it often enables the quantification of the amount of the substance under scrutiny. Both infrared and ultraviolet–visible spectroscopy are notably prevalent in analytical applications, exemplifying the versatility of absorption spectroscopy across diverse scientific disciplines.
The absorption intensity, varying with the frequency of electromagnetic radiation, gives rise to the absorption spectrum. This phenomenon is harnessed through absorption spectroscopy across the electromagnetic spectrum.
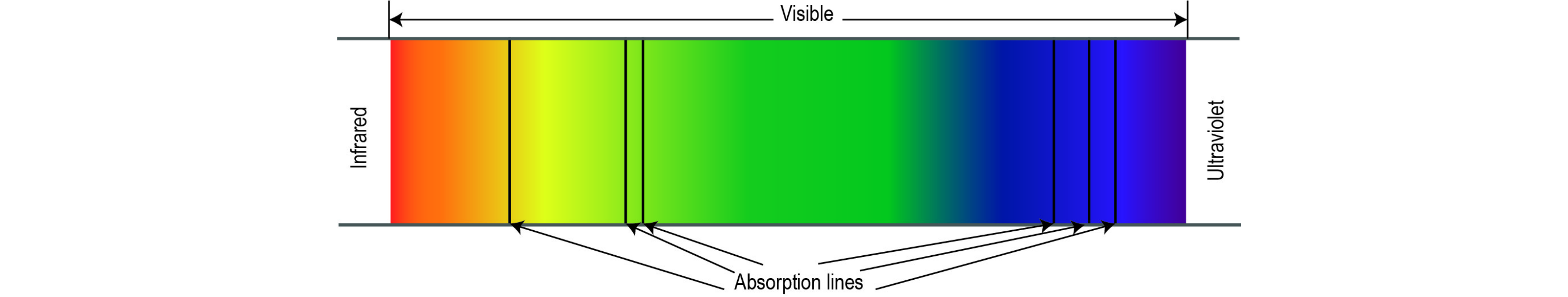
The utility of absorption spectroscopy in chemical analysis lies in its specificity and quantitative attributes. The specificity enables the differentiation of compounds within mixtures, making absorption spectroscopy applicable across a wide range of fields. Moreover, the specificity facilitates the identification of unknown samples by comparing measured spectra with a reference library. Even in cases where a library match is absent, qualitative information about a sample can be deduced. For example, infrared spectra exhibit characteristic absorption bands indicating the presence of specific bonds, such as carbon-hydrogen or carbon-oxygen. Recent technological strides in spectroscopy have enabled the discrimination of various isotopologues of CO2, treating them as distinct entities. CO2 isotopologues, resulting from the replacement of common isotopes 12C and 16O of carbon and oxygen with the less common 13C and 18O, exhibit unique absorption spectra in the mid-infrared range. The substitution of an atom by a heavier isotope, such as 13C replacing 12C or 18O replacing 16O, induces a spectrum shift, allowing for the differentiation between the two isotopologues. Key isotopologues of CO2 include 12C16O16O (mass 44), 13C16O16O (mass 45), and 12C16O18O (mass 46). Quantized rotational and vibrational states generate a distinctive spectral fingerprint. Determining the ratio of peaks from selected regions of the spectrum enables the accurate simultaneous measurement of 13C and 18O of CO2. Absorptions for CO2 are notably stronger in the mid-infrared compared to the near-infrared, allowing for the application of simple direct absorption approaches to achieve high specificity, sensitivity, and accuracy.
The differentiation of the three main CO2 isotopologues (with masses 44, 45, and 46) in the air is based on measurements of mid-infrared light absorption (λ ≈ 4.3 μm) and subsequent concentration calculation using the Lambert–Beer law. The analyzer collects measurements at 1 Hz, determining the 13C/12C ratio, 18O/16O ratios, and CO2 concentration. Laser-based analyzers offer advantages over more complex methods, such as IRMS, as CO2 sample purification is unnecessary, enabling high-frequency measurements of isotopic ratios. This method proves valuable in studying atmospheric CO2 patterns. Isotope analyzers automatically manage working standard gases for calibration, referencing measurements against isotopic working standards. Referencing is conducted hourly to achieve high precision and accuracy (σ = ± 0.25‰ for both δ13C and δ18O).
A calibration procedure correlates the analytical signal with the 13C/12C and 18O/16O ratios of working standards. Pure CO2 (purity >99.999 vol. %) with known 13C/12C and 18O/16O ratios against international standards serves as the reference.
For atmospheric gas collection, a stainless-steel capillary is utilized, fixed outside the Istituto Nazionale di Geofisica e Vulcanologia in Palermo (13.60 m above the ground floor).

Environmental variables, including air temperature, atmospheric pressure, relative humidity, rain rate, radiation, evapotranspiration, wind direction, and wind speed, are also measured. These variables play a crucial role in investigating pollutant patterns, especially regarding gas dispersion. Collected by a weather station on the roof of the ACO-Laboratory, the data avoids interference with surrounding buildings and provides insights into air circulation at the site of air CO2 monitoring. The station records all parameters with a 5-minute sampling frequency.
A thorough examination of the origins of gas emissions is attainable through the application of stable isotopic composition analysis, employing the Keeling plot approach. This methodology aids in understanding the sources contributing to atmospheric CO2 emissions by comparing isotopic data against the inverse of CO2 concentration. Rooted in a two-component mixing model, the Keeling plot presents a linear combination of isotopic mass balance, resulting in straight lines within the δ13C vs. 1/C plot (Keeling, 1958; Pataki et al., 2003). Both δ13C and δ18O play pivotal roles in distinguishing processes involving atmospheric CO2. While δ13C reveals information about CO2 sources, δ18O tracks CO2 fluxes through geospheres.
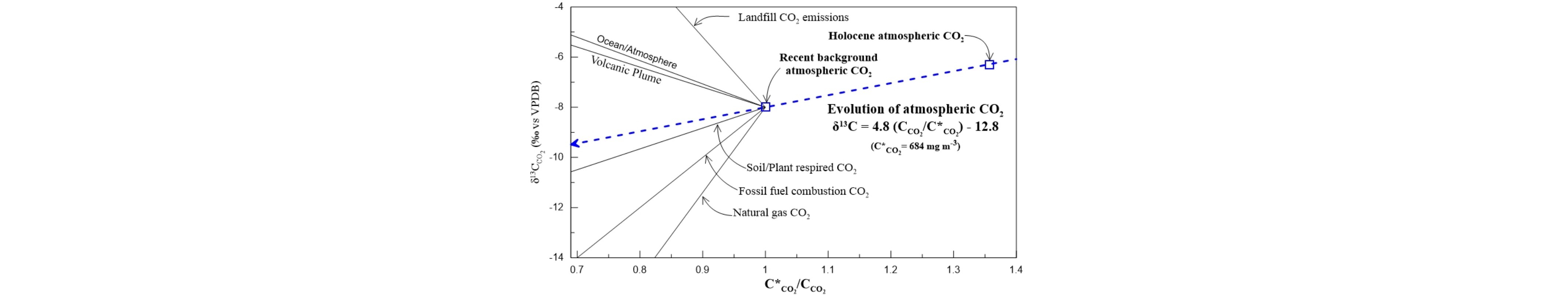
The keeling plot (see the above figure) shows the mixing lines between atmospheric CO2 and several sources of CO2 in the atmosphere either natural or anthropogenic. Line “Natural gas CO2” represents the addition of CO2 from combustion of natural gas (δ13C = -42‰ VPDB); Line “Fossil fuel combustion CO2” represents the addition of CO2 from combustion of petroleum (δ13C = -27‰ VPDB); Line “Soil/Plant respired CO2” represents the addition of CO2 from soil respiration (δ13C = -25.7‰ VPDB); Line “Volcanic Plume” represents the addition of CO2 from volcanic degassing (δ13C = +4.0‰ VPDB); Line “Ocean/Atmosphere” represents the addition of CO2 from ocean-atmosphere equilibrium (δ13C = +0.2‰ VPDB); Line “Landfill CO2 emissions” represents the addition of CO2 from waste decomposition (δ13C = +20.0‰ VPDB). Blue line shows the evolution of stable isotopes in CO2 and CO2 concentration between a Holocene pre-industrial atmospheric CO2 and the approximate global atmospheric CO2 at early XX1 century (380 ppm vol). For reference, the dashed line was extrapolated to CO2 concentrations higher than those of modern global atmospheric CO2.
The intercepts on the straight line in above figure indicates the carbon isotope composition of a variety of sources of CO2. The Keeling method offers insight into the isotopic signature of the CO2 source, under the assumption of constancy in both background and CO2 sources throughout the observation period. Urban zones, characterized by diverse human activities, present complex systems where various emissions contribute to the isotope composition in the atmosphere. In specific localities, geological outgassing, such as active or extinct volcanic degassing, emerges as the primary source of CO2 emissions.
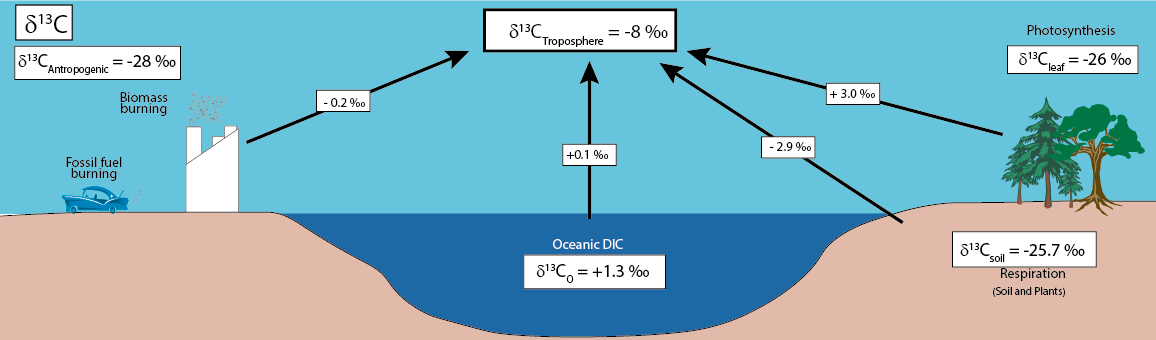
Annual mean forcings on the δ13C value of atmospheric CO2 (see the above figure after Yakir, 2003). Each specific forcing component represents the product of the involved flux, diluted by the atmospheric carbon pool, and the difference in δ13C values between the flux and the mean atmosphere. The values presented are order-of-magnitude estimates, as elaborated in the text, and entail notable uncertainties. The typical values for the relevant carbon reservoirs are provided in the accompanying boxes. Among these, the land biosphere exerts the most substantial forcing, driven by the contrasting impacts of photosynthesis and respiration. The net photosynthetic carbon sink tends to enrich the atmosphere in 13C. Comparatively, the oceanic forcing is nearly an order of magnitude smaller than the land effect, establishing a foundation for distinguishing between land and ocean sinks and sources. Fossil fuel emissions, conversely, act to dilute atmospheric 13C. Despite the nearly balanced nature of these distinct forcings, the fossil fuel effect emerges as the dominant contributor to atmospheric forcing, resulting in a net trend of 0.02‰ per year. The interplay of these forcing components showcases the complex dynamics within the carbon cycle, underscoring the importance of acknowledging the uncertainties inherent in such estimations. The δ18O signal demonstrates a unique coupling between the hydrological and carbon cycles, reflecting the exchange of CO2 with the terrestrial biosphere. The global δ18O background signal, established by atmosphere-ocean water exchange, undergoes modification on land through interactions with the biosphere (Ciais and Maijer, 1998). Oxygen atom replacement processes in atmospheric CO2 involve exchanges with water in seawater, soils, and plant leaves. CO2-H2O oxygen exchange in plant leaves, where carbonic anhydrase facilitates the equilibration of 18O content, transfers this signal to air CO2. The hydration of CO2 molecules, a crucial process for oxygen exchange, occurs slowly at 25 °C, primarily involving reactions with water and carbonic anhydrase in plant leaves (Mills and Urey, 1940). The isotopic equilibration does not occur in the atmosphere due to limited liquid water content, acting as a rate-limiting variable. Soil respiration, catalyzed by carbonic anhydrase in soils, introduces an alternative process affecting δ18O, with contrasting effects from photosynthesis.
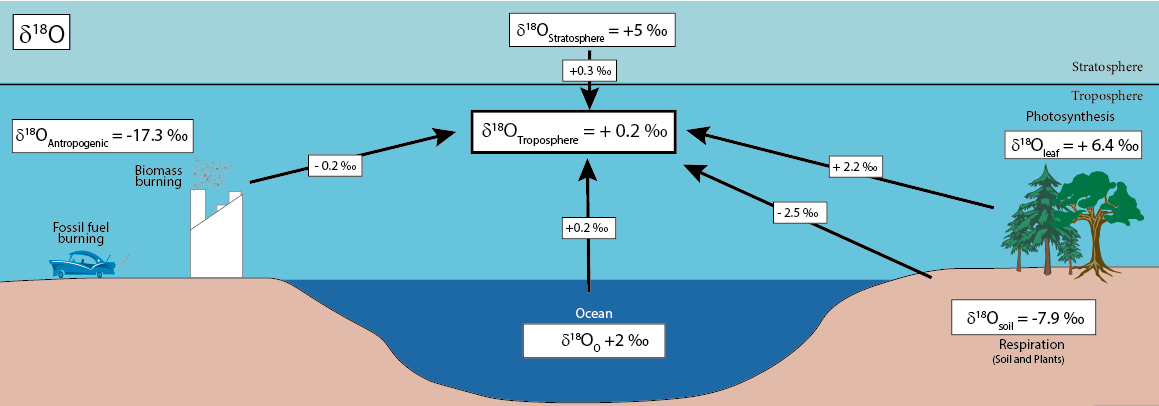
Annual mean "forcings" on the δ18O value of atmospheric CO2 delineate the impact of various influencing factors (see the above figure after Yakir, 2003). Each specific forcing component results from the product of the involved flux, diluted by the atmospheric carbon pool, and the difference in δ18O values between the flux and the mean atmosphere. The values presented are order-of-magnitude estimates, as detailed in the text, and are subject to significant uncertainties. The typical values for the involved reservoirs are provided in accompanying boxes, reflecting the δ18O value of the water in each reservoir. Notably, the anthropogenic reservoir is represented by the δ18O value of atmospheric oxygen.
Among these forcings, the land biosphere emerges as the most influential, marked by the contrasting effects of photosynthesis and respiration. Land photosynthesis tends to enrich the atmosphere in 18O, while soil respiration has the opposite effect. This dichotomy provides a foundation for distinguishing between the fluxes of photosynthesis and respiration. In contrast, the oceanic forcing is an order of magnitude smaller. Fossil fuel emissions, meanwhile, act to decrease 18O, yet this effect is often "washed away" by the substantial exchange of oxygen in CO2 with water. As a result, a discernible long-term trend in the atmospheric δ18O is not commonly observed. The intricate interplay of these forcings underscores the complexity of factors influencing the isotopic composition of atmospheric CO2, necessitating an acknowledgment of the inherent uncertainties in these estimations.
Interpreting δ18O can be complex, but frequent measurements with high precision offer insights into local CO2 source values. While uncertainties may arise, the Keeling plot method aids in retrieving δ18O, considering factors such as soil respiration and plant photosynthesis, providing valuable information about the local source's isotopic composition.